Das klinische Erscheinungsbild und die Ausprägung des Lymphödems variieren je nach dem beteiligten Gen und der vorliegenden Mutation. Die Formen des Lymphödems werden in fünf Kategorien unterteilt: (1) Erkrankungen mit somatischem Mosaik und segmentalen Wachstumsstörungen, (2a) syndromale Krankheitsbilder, (2b) Erkrankungen mit systemischer Beteiligung, (2c) kongenitale Lymphödeme und (2d) spät auftretende (late-onset) Lymphödeme nach dem ersten Lebensjahr. Die genetische Diagnostik erfolgt nach detaillierter Beurteilung der klinischen Symptome des Patienten und Einordnung in eine der oben genannten fünf Kategorien. Die Diagnostik erfolgt in der Regel gemäß einer Stufendiagnostik. Man beginnt typischerweise mit einer Basisdiagnostik, die zytogenetische und molekularzytogenetische Untersuchungen umfasst. Anschließend kann eine molekulargenetische Diagnostik mittels Einzelgen-Analysen, Gen-Panel-Untersuchungen, Exomsequenzierung oder Ganzgenomsequenzierung durchgeführt werden. Diese Untersuchungen ermöglichen die Identifizierung von genetischen Varianten oder Mutationen, die als Ursache für die vorliegende Symptomatik angesehen werden können. Die Kenntnis der ursächlichen, genetischen Veränderung ermöglicht in Verbindung mit einer humangenetischen Beratung Aussagen über den Vererbungsmodus, das Wiederholungsrisiko und mögliche Begleitsymptome. In vielen Fällen ist eine präzise molekulargenetische Diagnostik erforderlich, um die spezifische Form des primären Lymphödems eindeutig zu identifizieren.

Einleitung:
Aufbau und Entwicklung des Lymphgefäßsystems
Das Lymphgefäßsystem stellt neben dem Blutgefäßsystem eines der beiden Gefäßsysteme in höheren Wirbeltieren dar. Es erfüllt vielfältige Funktionen, darunter die Aufrechterhaltung der Gewebehomöostase, die Aufnahme von Nahrungsfetten sowie eine zentrale Funktion bei der Immunantwort. Lymphgefäße sind in nahezu allen vaskularisierten Geweben vorhanden und sind hierarchisch aufgebaut. Die kleinsten Lymphgefäße, auch initiale Lymphgefäße genannt, nehmen interstitielle Flüssigkeit und gelöste Stoffe sowie Zellen aus dem Zellzwischenraum auf, welche nach Aufnahme ins Gefäß als Lymphe bezeichnet wird.
Die Lymphe wird im Anschluss zu größeren Gefäßen, die als Präkollektoren und Kollektoren bezeichnet werden, transportiert, bevor die Lymphe über den Ductus thoracicus oder den Ductus lymphaticus dexter schließlich zurück in den Blutkreislauf geführt wird. Die initialen Lymphgefäße haben eine besondere Struktur mit eichenblattförmigen lymphatischen Endothelzellen (LEZs), die keine oder nur eine unvollständige Basalmembran aufweisen. Diese Zellen haben spezialisierte interzelluläre Verbindungen mit einer diskontinuierlichen Verteilung, die als „knopfartiges“ (button-like) Verteilungsmuster bezeichnet wird. Diese „Knöpfe“ fungieren wie primäre Klappen und ermöglichen den freien Eintritt von Flüssigkeit, Molekülen und Zellen zwischen benachbarten LEZs1.
Im Gegensatz dazu haben größer lumenisierte Präkollektoren und Kollektoren eine kontinuierliche Verteilung von Zell-Zell-Kontaktmolekülen, die als „zipper-like“ oder reißverschlussartig bezeichnet werden. Hierdurch wird das Austreten von Flüssigkeit aus dem Gefäß in das umgebende Gewebe verhindert. Diese Gefäße sind zudem teilweise mit glatten Muskelzellen und bikuspiden Klappen ausgestattet, die einen unidirektionalen Flüssigkeitstransport ermöglichen und den Rückfluss der Lymphe verhindern2. Die Wechselwirkung zwischen den Klappen und der sich zusammenziehenden glatten Muskulatur ermöglicht die Entstehung eines aktiven Transportmechanismus im Lymphgefäßsystem. Dieser Mechanismus wird durch die Bewegungen der umgebenden Skelettmuskulatur und Blutgefäße unterstützt.
Das lymphatische Gefäßsystem entsteht während der Embryonalentwicklung. Schon in frühen Entwicklungsstadien transdifferenzieren Subpopulationen von venösen Endothelzellen zu LEZs, indem sie den lymphatischen Transkriptionsfaktor Prospero homeobox protein 1 (PROX1) exprimieren und den vaskulären endothelialen Wachstumsfaktor-Rezeptor 3 (VEGFR‑3) hochregulieren. Diese Zellen verlassen im Anschluss ihre ursprünglichen venösen Gefäßbetten und bilden Zellstränge entlang eines VEGF-C-Gradienten, welcher der Ligand von VEGFR‑3 ist. Innerhalb von nur zwölf Stunden aggregieren diese und formen die ersten großlumigen Lymphgefäße, die als primordialer Ductus thoracicus (pTD) bekannt sind und oftmals auch als Lymphsack bezeichnet werden.
Neben dem venösen Ursprung der frühen LEZs konnten in Mäusen auch alternative zelluläre Ursprünge beschrieben werden. Ein Beispiel hierfür ist das blutbildende hämogene Endothel, das für die Entwicklung des mesenterialen Lymphgefäßsystems verantwortlich ist. Diese Vielfalt bei den Ursprüngen der LEZs trägt maßgeblich zur organspezifischen Entwicklung und Funktion des lymphatischen Gefäßsystems bei3.
Das Lymphgefäßsystem und Erkrankungen des Menschen
Im Gegensatz zum Blutgefäßsystem wurde das Lymphgefäßsystem lange Zeit nur begrenzt beachtet und seine Rolle in der Entstehung menschlicher Krankheiten folglich nur rudimentär erforscht. So wurde Anfang der 2000er Jahre dem Lymphgefäßsystem eine eher passive Rolle bei der Entstehung menschlicher Krankheiten zugeschrieben. Neuere Untersuchungen haben jedoch dieses vorherrschende Dogma aufgebrochen und konnten dem Lymphgefäßsystem eine Schlüsselrolle bei zahlreichen Pathologien zuordnen. Hierzu zählen nicht nur lymphgefäßspezifische Erkrankungen wie das Lymphödem, sondern auch die vier großen medizinischen Herausforderungen des 21. Jahrhunderts: Krebs, Herz-Kreislauf-Erkrankungen, Immunität und Infektion sowie Adipositas4.
In Übereinstimmung mit seiner physiologischen Rolle manifestiert sich eine Störung des Lymphgefäßsystems klinisch durch drei Hauptsymptome: Erstens führt sie zu einer Beeinträchtigung der Gewebeimmunität, was wiederum rezidivierende oder anhaltende Infektionen begünstigt. Zweitens führt sie zu einer veränderten Aufnahme von Fetten im Darm. Drittens verändert sie die Gewebsflüssigkeitshomöostase, wodurch es zur Ausbildung von Lymphödemen kommt.
Gemäß dem überarbeiteten Sterling-Prinzip erfolgt die Aufnahme nahezu sämtlicher extravasalen Flüssigkeit sowie der darin enthaltenen Proteine aus dem interstitiellen Raum durch das Lymphgefäßsystem. Nur geringe Mengen werden temporär über die direkte venös-kapilläre Reabsorption dem System zurückgeführt. Dementsprechend sind sämtliche chronischen peripheren Ödeme entweder auf ein absolutes Versagen des Lymphtransports zurückzuführen oder auf eine hohe Lymphlast, die die Lymphdrainagekapazität übersteigt5.
Zusätzlich zur Schwellung kann ein Lymphödem auch durch Fibrosierung, chronische Entzündungen und abnormale Fettablagerungen gekennzeichnet sein. Neben den medizinischen Aspekten kann ein chronisches Lymphödem die Lebensqualität der Patienten erheblich beeinträchtigen, Beschwerden verursachen und die Betroffenen bei alltäglichen Aufgaben einschränken6.
Das Lymphödem
Lymphödeme lassen sich in zwei Kategorien unterteilen: primäre und sekundäre Lymphödeme. Ein primäres Lymphödem resultiert aus angeborenen Fehlbildungen, Funktionsstörungen oder einer angeborenen „Schwäche“ des Lymphgefäßsystems. Im Gegensatz dazu wird ein sekundäres Lymphödem durch eine Schädigung des Lymphgefäßsystems ausgelöst, wie sie beispielsweise als Folge eines Traumas oder einer onkologischen Behandlung auftreten kann.
Klinisch äußern sich periphere Lymphödeme durch Schwellungen im Gesicht, Rumpf, Genitalbereich oder an den Extremitäten, wobei die unteren Extremitäten am häufigsten betroffen sind. Es gibt jedoch auch systemische Manifestationen von Lymphödemen, die sich in Form von Hydrops fetalis, Aszites, intestinalen und pleuralen Lymphangiektasien, Chylothorax sowie Pleura- und Perikardergüssen äußern können7 8.
Der Hydrops fetalis ist kein eigenständiges Krankheitsbild. Es stellt ein Symptom dar, das bei verschiedenen Erkrankungen auftreten kann. Es äußert sich durch eine generalisierte Ansammlung von Flüssigkeiten in den serösen Körperhöhlen und Weichteilen des Fötus. Je nach Ursache kann zwischen immune und non-immune Hydrops fetalis unterschieden werden. Der immune Hydrops fetalis kann beispielsweise bei Rhesus-Inkompatibilität oder Myasthenie auftritt, der non-immune Hydrops fetalis kann idiopathisch oder auf multifaktorielle Ursachen zurückzuführen sein. Daneben kann er auch als Symptom im Rahmen eines genetisch bedingten Syndroms auftreten und folglich bereits ein erster Hinweis auf das Vorliegen eines primären Lymphödems sein9.
Das primäre Lymphödem – gegenwärtige Klassifikation
Die aktuelle Klassifikation des primären Lymphödems basiert auf einem Algorithmus, der 2010 am St. George‘s Hospital in London eingeführt wurde10. Diese kontinuierlich weiterentwickelte Klassifikation stellt eine wesentliche Erweiterung gegenüber der früheren Klassifikation des Lymphödems dar, die nicht mehr aktuell ist und daher nicht mehr angewandt werden sollte. Die ursprüngliche Klassifikation unterschied zwischen kongenitalen Lymphödemen, Lymphödemen während der Pubertät (Lymphoedema praecox) und Lymphödemen nach dem 35. Lebensjahr (Lymphoedema tarda).
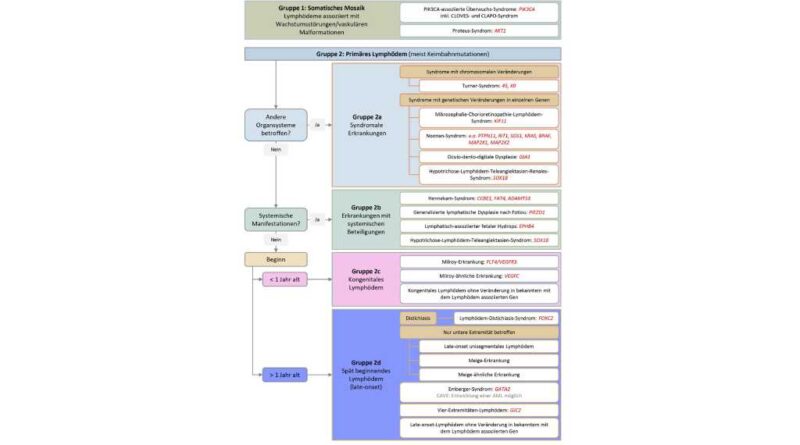
Durch neuere Untersuchungen zur Genetik, Klinik und Histologie des primären Lymphödems konnte jedoch gezeigt werden, dass die ursprüngliche Klassifikation zu vereinfacht war und den klinischen Phänotyp nicht ausreichend berücksichtigte. Infolgedessen unterscheidet der ursprünglich von Connell et al. entwickelte Algorithmus (in der gegenwärtigen Version von Gordon et al.11, die verschiedenen Formen des primären Lymphödems anhand klinischer Kriterien (Abb. 1).
Dieser Algorithmus unterscheidet fünf Gruppen12 13 14 15 16:
- angeborene Lymphödeme, die mit Gefäßmalformationen oder segmentalen Wachstumsstörungen assoziiert sind (somatische Mosaike).
- primäre Lymphödeme, die aufgrund von Keimbahnveränderungen auftreten:
a. primäre Lymphödeme im Rahmen einer übergeordneten syndromalen Erkrankung
b. primäre Lymphödeme mit systemischen Beteiligungen
c. kongenitale primäre Lymphödeme, die bereits vor der Geburt oder innerhalb der ersten zwölf Lebensmonate auftreten
d. später auftretende primäre Lymphödeme (late-onset Lymphödeme)
Krankheiten, die mit segmentalen Wachstumsstörungen wie Großwuchssyndromen und Gefäßmalformationen in Verbindung stehen, werden häufig auf das Vorliegen somatischer Mosaike zurückgeführt. Bei solchen Mosaiken weisen nicht alle Zellen des Körpers die genetische Veränderung auf. Stattdessen sind diese Veränderungen auf Zellen in der betroffenen Region oder im betroffenen Gewebe beschränkt. Dies ist darin begründet, dass die zugrunde liegende genetische Mutation nicht bereits bei der Befruchtung der Eizelle durch das Spermium auftritt, sondern erst zu einem späteren Zeitpunkt während der Embryonalentwicklung. Daher sind die genetischen Veränderungen nur in den Zellen vorhanden, die von der mutierten Zelle abstammen und sich aus ihr entwickeln.
In der Regel führt eine solche somatische Veränderung zu übermäßigem Wachstum des betroffenen Gewebes, das entweder aus den mutierten Zellen hervorgegangen ist oder von ihnen beeinflusst wird. Der Ort und der Zeitpunkt der somatischen Veränderung während der Embryonalentwicklung beeinflussen das Muster der auftretenden Symptome.
Neben dem Lymphgefäßsystem können auch Blutgefäße, Bindegewebe, Knochen oder Fettgewebe betroffen sein17 18. Häufig zeigt sich eine Aktivierung des PI3K-AKT-mTOR-Signalwegs, was zu gesteigerter Zellproliferation, Zellwachstum und Zellüberleben führt. Somatische Mutationen im PIK3CA-Gen, das für die katalytische Untereinheit der Phosphatidylinositol-3-Kinase (PI3K) kodiert, sind mit übermäßigem Wachstumssyndromen assoziiert, wie dem CLOVE-Syndrom (Congenital lipomatous overgrowth, vascular malformations, and epidermal nevi), das auch das Skelettsystem betreffen kann (CLOVES-Syndrom, OMIM #612918), und dem CLAPO-Syndrom (Capillary malformation of the lower lip, lymphatic malformation of the face and neck, asymmetry of the face and limbs, and partial/generalized over-growth, OMIM #613089). Darüber hinaus können aktivierende somatische Veränderungen im AKT1-Gen nachgewiesen werden, die mit dem Proteus-Syndrom in Verbindung stehen (OMIM #176920). Dieses Syndrom zeigt ein vielfältiges Krankheitsbild mit asymmetrischem und übermäßigem Wachstum von Körperteilen, Dysregulation des Fettgewebes und Gefäßfehlbildungen19 20 21 22.
Diagnostik somatischer Mosaike
Da somatische Veränderungen nicht alle Zellen im menschlichen Körper betreffen, erfolgt der Nachweis nicht über das Blut, sondern durch die Analyse von DNA aus dem betroffenen Gewebe. Eine alternative Methode, den Nachweis aus dem Blut durchzuführen, stellt die Liquid Biopsy dar. Obwohl dies derzeit noch nicht als diagnostischer Standard etabliert ist, birgt es das Potenzial, in der Zukunft somatische Mosaike auch bei Patienten zu identifizieren, bei denen keine Gewebebiopsie möglich ist.
Gruppe 2:
Primäre Lymphödeme ohne Assoziation mit Überwuchs oder Gefäßmalformationen
Ursächlich für das primäre Lymphödem sind häufig Veränderungen in Genen, die eine entscheidende Rolle bei der Entwicklung und Funktion der Lymphgefäße spielen. So resultieren diese Mutationen oft in Fehlanlagen oder Funktionsbeeinträchtigung der Lymphgefäße und Lymphgefäßklappen. Diese Mutationen liegen in der Regel als Keimbahnveränderungen vor, die entweder vererbt oder beim Betroffenen neu aufgetreten sind (de novo) (Tab. 1).
Für die molekulargenetische Diagnostik kann DNA aus einer Blutprobe genutzt werden. Da die zugrunde liegende genetische Veränderung alle Zellen im Körper betrifft, kann hier das Blut als Stellvertretergewebe für alle Körperzellen einschließlich der Lymphgefäße genutzt werden.
Gruppe 2a:
Primäre Lymphödeme im Rahmen einer syndromalen Erkrankung
Innerhalb der Gruppe der primären Lymphödeme, die im Kontext syndromaler Erkrankungen auftreten, werden in erster Linie Syndrome berücksichtigt, die mit strukturellen Veränderungen der Chromosomen oder Mutationen in einzelnen Genen einhergehen. Obwohl das primäre Lymphödem bei dieser Gruppe als eines der Symptome auftritt, ist es nicht das dominierende phänotypische Merkmal23.
Eine häufige Ursache für ein primäres Lymphödem, das mit numerischen oder strukturellen Veränderungen der Chromosomen einhergeht, ist das Turner-Syndrom. Bei vielen Schwangerschaften mit Turner-Syndrom kommt es bereits vor der Geburt zu schwerwiegendem Hydrops fetalis, was zu einer Fehlgeburt führen kann. Bei lebend geborenen Kindern mit Turner-Syndrom konnte pränatal oft auch ein Hydrops fetalis beobachtet werden, der sich jedoch im weiteren Verlauf der Schwangerschaft größtenteils zurückbildet. Nach der Geburt sind die hauptsächlichen phänotypischen Merkmale meist nur noch eine beidseitige Hautfalte am Hals zwischen Mastoid und Akromion, bekannt als Pterygium colli, sowie Lymphödeme an Händen und Fußrücken. Die betroffenen Individuen weisen einen weiblichen Phänotyp auf. Neben dem lymphatischen Phänotyp, der nur ein Teilsymptom des variabel ausgeprägten Turner-Syndroms darstellt, liegen häufig auch angeborene Herzfehler, Kleinwuchs und Gonadendysgenesie vor24 25.
Diagnostik bei chromosomalen Veränderungen
Das Turner-Syndrom ist normalerweise durch den Karyotyp 45,X charakterisiert, es können jedoch auch verwandte gonosomale Mosaiken wie 45,X/46,XX oder 45,X/46,XY auftreten. Bei Vorliegen eines Mosaiks mit einer 46,XY-Zelllinie besteht ein erhöhtes Risiko für maligne Keimzelltumore. Daher sollte der zugrunde liegende Karyotyp durch eine Chromosomenanalyse einschließlich Interphase FISH (Fluoreszenz-in-situ-Hybridisierung) aus Lithium-Heparin-Blut bestimmt werden.
Im Gegensatz zu chromosomalen Veränderungen können primäre Lymphödeme dieser Gruppe auch durch Veränderungen in einzelnen Genen verursacht werden. Die Diagnostik erfolgt entsprechend einem festgelegten Stufenschema.
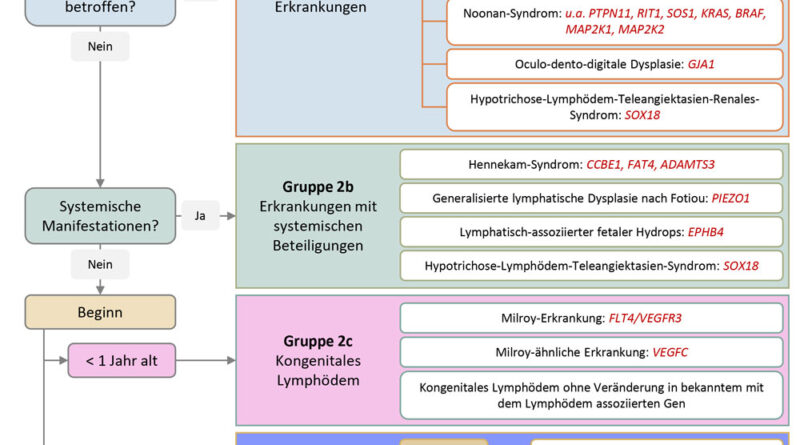
Das Noonan-Syndrom (OMIM #163950) ist ein Beispiel für ein primäres Lymphödem, das im Kontext syndromaler Erkrankungen auftritt. Es ist durch postnatalen Kleinwuchs, charakteristische Dysmorphiezeichen des Gesichts, angeborene Herzfehler und zum Teil auch Entwicklungsverzögerungen gekennzeichnet. Das Noonan-Syndrom zählt zu den RASopathie-Syndromen, bei denen Mutationen in Genen des RAS-Signalwegs vorliegen26 27.
Das Mikrozephalie-Chorioretinopathie-Lymphödem-Syndrom (OMIM #152950) ist gekennzeichnet durch Gehirnfehlbildungen einschließlich Intelligenzminderung, Chorioretinopathie und ein primäres Lymphödem der unteren Extremitäten. Die genetische Ursache ist eine Veränderung im KIF11-Gen28.
Die okulo-dento-digitale Dysplasie (OMIM #164200) betrifft vor allem Augen, Zähne und Finger. Mikrophthalmie, Zahnanomalien und Fingerfehlbildungen sind charakteristisch, und ein primäres Lymphödem ist in der Regel vorhanden. Die Ursache sind Veränderungen im GJA1-Gen, welches für das Protein Connexin 43 kodiert. Connexine sind essentiell für die Zell-Zell-Kommunikation und funktionelle Ausbildung von Lymphgefäßklappen29 30.
Das Emberger-Syndrom (OMIM #614038) stellt ein Late-onset-Lymphödem dar, bei dem v. a. ein Lymphödem der unteren Extremität und der Genitalien vorliegt. Da es bei Vorliegen eines Emberger-Syndroms auch zur Ausbildung einer Myelodysplasie und akuter myeloischer Leukämie kommen kann, sollte grundsätzlich bei Kindern mit primären Lymphödem eine molekulargenetische Untersuchung auf das Vorliegen des Emberger-Syndroms durchgeführt werden. Ursächliche Veränderungen finden sich im Gen für den Transkriptionsfaktor GATA2, der eine wichtige Rolle in hämatopoetischen Stammzellen und Endothelzellen spielt. Die engmaschige Überwachung ist daher bei Verdacht auf das Emberger-Syndrom besonders wichtig31.
Gruppe 2b:
Primäre Lymphödeme mit systemischer Beteiligung
In der Gruppe 2b werden Erkrankungen mit systemischer Beteiligung zusammengefasst, bei denen das primäre Lymphödem das Leitsymptom ist.
Das Hennekam-Syndrom stellt ein progredientes kongenitales primäres Lymphödem dar, das den gesamten Körper betreffen kann. Es geht mit fazialen Dysmorphiezeichen und systemischen Manifestationen wie Hydrops fetalis, intestinalen Lymphangiektasien, Chylothorax und Aszites einher. Das Hennekam-Syndrom wird durch genetische Veränderungen in den Genen CCBE1 (Hennekam-Syndrom Typ 1, OMIM #235510), FAT4 (Hennekam-Syndrom Typ 2, OMIM #616006) oder ADAMTS3 (Hennekam-Syndrom Typ 3, OMIM #618154) verursacht. Mutationen in CCBE1 und ADAMTS3 führen zu einer reduzierten Prozessierung des lymphatischen vaskulären Wachstumsfaktors VEGF‑C, was zu einer Fehlentwicklung der Lymphgefäße während der Embryonalentwicklung führt. Im Gegensatz dazu beeinflussen pathogene Veränderungen im FAT4-Gen die Bildung der Lymphgefäßklappen und erklären die phänotypischen Unterschiede zwischen den Hennekam-Unterformen32 33 34.
Die generalisierte lymphatische Dysplasie nach Fotiou (OMIM #616843) zeigt ähnliche Merkmale wie das Hennekam-Syndrom, wobei das primäre Lymphödem weniger stark ausgeprägt ist. Diese Erkrankung wird durch pathogene Veränderungen im PIEZO1-Gen verursacht, das für einen mechanosensitiven Ionenkanal kodiert, der für die Mechanosignalübertragung während der Lymphangiogenese essenziell ist35.
Das Hypotrichose-Lymphödem-Teleangiektasien-Syndrom (OMIM #607823) ist hauptsächlich mit einem Lymphödem der unteren Extremitäten assoziiert. Systemische Symptome umfassen Hydrops fetalis und bei männlichen Betroffenen eine Hydrozele. Teleangiektasien sind ein weiteres dermatologisches Merkmal. Bei Vorliegen eines zusätzlichen renalen Phänotyps wird dies als Hypotrichose-Lymphödem-Teleangiektasien-Renales Syndrom (HLTRS, OMIM#137940) bezeichnet. Ursächlich sind pathogene Veränderungen im SOX18-Gen, einem Transkriptionsfaktor, der während der Embryonalentwicklung die Expression des lymphatischen Transkriptionsfaktors PROX1 in venösen Endothelzellen induziert. Pathogene Veränderungen in SOX18 führen zu einer veränderten PROX1-Expression, einer verminderten Migration von lymphatischen Endothelzellen und damit zur Fehlbildung des Lymphgefäßsystems36 37.
Gruppe 2c:
Kongenitale primäre Lymphödeme
Die häufigste Form des primären Lymphödems ist die Milroy-Erkrankung (OMIM #153100). Sie zeigt das typische Erscheinungsbild eines kongenitalen Lymphödems, das in der Regel bilateral in den unteren Extremitäten auftritt, insbesondere unterhalb der Knie und auf dem Fußrücken. Zusätzlich sind häufig Varikosen an den Beinen zu beobachten, und männliche Betroffene können eine Hydrozele entwickeln. Die zugrunde liegenden pathogenen Veränderungen wurden im FLT4-Gen identifiziert. FLT4 kodiert für den vaskulären endothelialen Wachstumsfaktor-Rezeptor 3 (VEGFR‑3), der im Wesentlichen in Lymphgefäßen exprimiert wird. Er spielt eine entscheidende Rolle bei der Entwicklung und Funktion dieser Gefäße. VEGFR‑3 wird von LEZs exprimiert und bindet den Wachstumsfaktor VEGF‑C. Diese Bindung führt zur Aktivierung des Rezeptors und fördert Migration, Proliferation und Wachstum der LEZs38.
Seltener werden Veränderungen im Gen für den Liganden VEGF‑C gefunden, was zu einer Milroy-ähnlichen Erkrankung (OMIM #615907) führt. Klinisch ähneln sich beide Formen, daher ist eine eindeutige Diagnose und Klassifizierung nur durch eine molekulargenetische Analyse möglich. In der Regel werden diese Erkrankungen vererbt, obwohl auch sporadische Fälle ohne familiäre Vorgeschichte auftreten können (de novo) 39.
Daneben gibt es auch weitere Formen des kongenitalen Lymphödems, wie z. B. multisegmentale und unilaterale Ödeme sowie Lymphödeme mit Genitalbeteiligung. Die zugrunde liegenden Gene für diese Erkrankungen sind jedoch bisher unbekannt40 41.
Gruppe 2d:
Spätmanifestierende primäre Lymphödeme
Das Lymphödem-Distichiasis-Syndrom (OMIM #153400) repräsentiert eine Form des late-onset primären Lymphödems. Dieses Lymphödem, das in der Regel während der Pubertät auftritt, zeichnet sich durch das Vorhandensein einer Distichiasis aus. Distichiasis ist das Auftreten einer zweiten Reihe von Wimpern oder nur einzelner Wimpern, die aus den Meibom‘schen Drüsen auf der Innenseite des Augenlids auswachsen. Die Verdachtsdiagnose kann durch gezielte Anamnese und Untersuchung des Betroffenen auf das Vorliegen einer Distichiasis gestellt werden und die molekulargenetische Analyse bestätigt die Diagnose. Pathogene Veränderungen im Gen FOXC2 sind die Ursache für das Lymphödem-Distichiasis-Syndrom. FOXC2 spielt eine bedeutende Rolle bei der Entwicklung, Funktion und Aufrechterhaltung der Lymphgefäßklappen und gewährleistet somit den unidirektionalen Transport der Lymphe. Da Lymphgefäß- und Venenklappen viele gemeinsame Eigenschaften in Bezug auf Entwicklung und Signalwege aufweisen, führt eine Mutation in FOXC2 zu einer verstärkten Dysfunktion der Lymphgefäß- und Venenklappen, was wiederum das Lymphödem und die Varikose verursacht42.
Die Meige-Erkrankung (OMIM #153200) ist eine weitere Form des spät manifestierenden primären Lymphödems. Dieses Lymphödem tritt während der Pubertät oder im Erwachsenenalter auf und betrifft typischerweise die untere Extremität. In der Regel handelt es sich um ein isoliertes Lymphödem, ohne das Vorliegen von Varikosen. Obwohl familiäre Häufungen bekannt sind, wurde bisher kein assoziiertes Gen identifiziert43.
Das Vier-Extremitäten-Lymphödem (OMIM #613480) tritt hauptsächlich während der Adoleszenz auf. Es beginnt in der Regel in der unteren Extremität und breitet sich später auf die obere Extremität aus. Ursächlich sind pathogene Veränderungen im Gen GJC2, das für das Gap-Junction-Protein Connexin 47 kodiert. Wie die meisten Connexine spielt auch Connexin 47 eine zentrale Rolle bei der Bildung von Gap Junctions, die für die Zell-Zell-Kommunikation zwischen den Endothelzellen der Lymphgefäßklappen entscheidend sind. Daher führen Mutationen in diesem Gen zu einer Dysfunktion oder Fehlentwicklung der Gefäßklappen44 45.
Neben den hier beschriebenen Formen des primären Lymphödems und den jeweils assoziierten Genen konnten zwischenzeitig auch weitere Gene als Ursache für angeborene Erkrankungen des Lymphgefäßsystems identifiziert wurden. Aufgrund zunehmender genetischer Testung und Forschung im Bereich der Lymphgefäß-Genetik ist zu erwarten, dass die Anzahl der mit diesen Erkrankungen assoziierten Gene in Zukunft weiter zunehmen wird und in den bestehenden Diagnosealgorithmus integriert wird. Als Beispiel sei hier das Gen CELSR1 genannt, das eine Schlüsselrolle im Planar-Cell-Polarity-Signalweg spielt. Veränderungen in CELSR1 sind mit angeborenen bilateralen, spät auftretenden Lymphödemen der unteren Extremität assoziiert. Es wurden auch renale Veränderungen beschrieben, daher wird bei betroffenen Personen eine nephrologische Untersuchung empfohlen. Die Vererbung erfolgt autosomal-dominant mit variabler Expressivität und inkompletter Penetranz, die bei weiblichen Trägern bei 91 % und bei männlichen Trägern bei 23 % variiert46.
Durchführung genetischer Diagnostik bei primären Lymphödemen
Die zielgerichtete molekulargenetische Diagnostik von primären Lymphödemen erfordert neben der klinischen Untersuchung auch eine sorgfältige Familienanamnese, um eine Zuordnung zu einer der fünf Gruppen zu ermöglichen. Im Gegensatz zu Erkrankungen der Gruppe 1, bei denen somatische Veränderungen im betroffenen Gewebe vorliegen und daher eine Untersuchung des betroffenen Gewebes erfordern, wird bei den primären Lymphödemen der Gruppe 2 in der Regel von einer Keimbahnveränderung ausgegangen. Daher reicht in der Regel eine Untersuchung von Blut aus, das als repräsentatives Organ für alle Körperzellen betrachtet wird.
Für die Diagnosestellung stehen verschiedene Methoden zur Verfügung, darunter die konventionelle Zytogenetik (Chromosomenanalyse), die Molekularzytogenetik (Array-CGH [Array-basierte Comparative Genom Hybridisierung] und FISH) sowie die Molekulargenetik (Sequenzanalyse, quantitative PCR und MLPA [Multiplex Ligation-dependent Probe Amplification]).
Die Auswahl der geeigneten Methode hängt von der spezifischen Verdachtsdiagnose ab. Zum Beispiel wird bei Verdacht auf das Vorliegen eines Turner-Syndroms eine Chromosomenanalyse durchgeführt.
Molekulargenetische Stufendiagnostik des primären Lymphödems
Die molekulargenetische Testung auf Vorliegen eines primären Lymphödems erfolgt in der Regel an EDTA-Blutproben, da die meisten dieser Erkrankungen auf genetische Veränderungen in einzelnen Genen zurückzuführen sind. Diese Untersuchung zielt darauf ab, kleinere Deletionen oder Punktmutationen zu identifizieren, das heißt Veränderungen einzelner oder weniger benachbarter Basenpaare im jeweiligen Gen. Dabei kann die Untersuchung einem Stufenschema folgen:
Wenn ein starker klinischer Verdacht auf das Vorliegen einer bestimmten Form des Lymphödems besteht, z. B. das Lymphödem-Distichiasis-Syndrom bei Vorliegen einer Distichiasis, kann eine gezielte Einzelgen-Untersuchung durchgeführt werden.
Ist kein spezifischer Verdacht vorhanden, werden durch den Einsatz der Hochdurchsatz-Sequenzierungstechnik (Next Generation Sequencing, NGS) nicht nur einzelne, sondern mehrere Gene gleichzeitig sequenziert (Multi-Gen-Analyse, Paneldiagnostik).
Die umfangreichste Form der Paneldiagnostik ist die Exomsequenzierung, bei der alle protein-kodierenden Bereiche und flankierenden Intronbereiche aller bekannten proteinkodierenden menschlichen Gene analysiert werden.
Die Exomsequenzierung eignet sich besonders gut für unklare syndromale Erkrankungen mit Verdacht auf eine seltene Ursache. Sie bietet auch die Möglichkeit, neue Kandidatengene zu identifizieren, die zuvor nicht mit der vorliegenden Symptomatik in Verbindung gebracht wurden. Wenn eine positive Familienanamnese vorliegt und insbesondere Kinder betroffen sind, kann die sogenannte Trio-Exomsequenzierung in Erwägung gezogen werden. Dabei werden Proben sowohl vom Betroffenen als auch von den Eltern sequenziert, um De-novo-Mutationen zu diagnostizieren, die erstmals beim Betroffenen aufgetreten sind. Dieses Verfahren kann auch bei vermutetem autosomal-rezessivem Erbgang das biallelische Vererbungsmuster bestätigen.
Die umfassendste Form der Sequenzanalyse ist die Ganzgenomsequenzierung. Derzeit wird dieses Verfahren jedoch nur von wenigen humangenetischen Instituten im Rahmen der Routinediagnostik durchgeführt, sodass gegenwärtig Ganzgenomsequenzierungen oft nur in Forschungsstudien eingesetzt werden.
Diese verschiedenen molekulargenetischen Untersuchungsmethoden tragen dazu bei, die genetischen Ursachen von primären Lymphödemen genau zu identifizieren und die Diagnose präzise zu stellen.
Histologische Diagnostik
Die histologische Analyse des betroffenen Gewebes ist eine zusätzliche diagnostische Methode, die zur Untersuchung von primären Lymphödemen eingesetzt werden kann. Die klassische zweidimensionale Histologie ist derzeit der Goldstandard in der Diagnostik. Allerdings kann diese Methode aufgrund ihrer Begrenzung auf zweidimensionale Schnittbilder die komplexe Gefäßarchitektur möglicherweise nicht ausreichend detailliert beschreiben.
Um eine umfassendere Analyse zu ermöglichen, wird die Verwendung einer dreidimensionalen Histologie der gesamten Probe empfohlen, die auf Lichtblattmikroskopie-basierter Schnittbildgebung beruht. Diese Technik wird derzeit hauptsächlich in klinischen Studien und Forschungsprojekten eingesetzt. Sie ermöglicht es, nach vorheriger Immunfluoreszenzfärbung relevanter Strukturen wie Lymph- und Blutgefäßen die gesamte Probe mit Einzelzellauflösung optisch zu schneiden und anschließend die dreidimensionale Gefäßarchitektur zu rekonstruieren. Dieser Ansatz erlaubt es, Gefäßveränderungen im räumlichen Kontext zu identifizieren.
Die 3D-Histologie hat nicht nur diagnostische, sondern auch klinische Relevanz. Sie kann als Entscheidungshilfe bei der Auswahl möglicher pharmakologischer und chirurgischer Interventionen dienen. Durch den Vergleich des Phänotyps mit Referenzdaten können Ärzte und Forscher wichtige Einblicke in die Pathophysiologie der Erkrankung gewinnen und geeignete Behandlungsstrategien entwickeln47 48.
Fazit
Angeborene Lymphödeme stellen eine Form der Fehlentwicklung bzw. Dysfunktion von Lymphgefäßen dar, die sich bereits embryonal ausbildet. Aufgrund der Vielzahl an Molekülen, die an der Entwicklung der Lymphgefäße beteiligt sind, sowie der komplexen Gefäßarchitektur und den Gefäßbett-abhängigen Funktionen können die Ausprägungen, der Schweregrad sowie Symptome eines Lymphödems sehr variieren. Entsprechend der Ursache und Klinik werden primäre Lymphödeme in fünf Kategorien unterteilt, auf deren Basis eine molekulargenetische Diagnose gestellt werden kann. Humangenetische Testungen auf das Vorliegen eines primären Lymph-ödems sollten in einem spezialisierten Zentrum erfolgen, um eine möglichst breite Analyse durchführen zu können. Die molekulargenetische Analyse kann mit räumlicher 3D-Histologie kombiniert werden, um den Genotyp mit dem resultierten Phänotyp zu korrelieren.
Der Autor
Dr. rer. nat. Dr. med. René Hägerling
Lymphovaskuläre Medizin und Translationale 3D-Histologie,
Institut für Medizinische Genetik und Humangenetik
Charité – Universitätsmedizin Berlin
Augustenburger Platz 1
13353 Berlin
Rene.Haegerling@charite.de
Begutachteter Beitrag/reviewed paper
Hägerling R. Die Genetik und Diagnostik des primären Lymphödems. Orthopädie Technik, 2023; 74 (11): 28–37
Tab. 1 Gene und assoziierte Krankheitsbilder. Dargestellt sind die Gene und assoziierten Krankheitsbilder, die als ursächlich für ein primäres Lymphödem in Betracht gezogen werden können. Eine Übersicht der typischen Klinik der Betroffenen und der Funktion des Genprodukts sind dargestellt (modifiziert nach Ott C‑E, Danyel M, Kemper C, Hägerling R. Genetische Diagnostik des primären Lymphödems. Lymphologie in Forschung und Praxis, 2021; 25 (1) :14–20).
| Tabelle 1 | ||||||
|---|---|---|---|---|---|---|
| Gen | Krankheitsbild | OMIM-Nummer | Klinik | Funktion des Genprodukts | ||
| GATA2 | Emberger Syndrom | 614038 | Syndromale Erkrankungen | - Lymphödem der unteren Extremität und Genitalien | Transkriptionsfaktor, der in hämatopoetischen Stammzellen und Endothelzellen exprimiert | |
| (Gruppe 2a) | - Myelodysplasie bis AML | |||||
| KIF11 | Mikrozephalie-Chorioretinopathie-Lymphödem Syndrom | 152950 | - Lymphödem der unteren Extremität | Motorprotein, das eine Rolle in Mitose und Vesikeltransport spielt | ||
| - Intelligenzminderung | ||||||
| - Chorioretinopathie | ||||||
| GJA1 | Oculo-dento-digitales Syndrom | 164200 | - Lymphödem | Connexin 43 spielt als Bestandteil von Gap-Junctions in Lymphgefäß-Klappen eine Rolle in der korrekten Ausbildung ebendieser | ||
| - Mikroophthalmie | ||||||
| - Kleine oder fehlende Zähne | ||||||
| - Schwacher Zahnschmelz | ||||||
| - Camptodaktylie | ||||||
| - Syndaktylie zw. vierten und fünften Finger (evtl. auch der Zehen) | ||||||
| CCBE1 | Hennekam-Syndrom | Typ 1 | 235510 | Lymphödem mit systemischer Beteiligung (Gruppe 2b) | - Progredientes, kongenitales primäres Lymphödem den gesamten Körper betreffend | Spielt eine Rolle in der Prozessierung von VEGF‑C (lymphatischer Wachstumsfaktor) und so in der Migration der Lymph-Endothelzellen (LEZs) und der Proliferation der Lymphgefäße |
| - Intestinale Lymphangiektasien | ||||||
| - Hydrops fetalis | ||||||
| - Chylothorax | ||||||
| ADAMTS3 | Typ 3 | 618154 | - Aszites | |||
| FAT4 | Typ 2 | 616006 | - Fasziale Auffälligkeiten | Spielt eine Rolle in Ausbildung der Lymphgefäßklappen | ||
| PIEZO1 | Generalisierte lymphatische Dysplasie nach Fotiou | 616843 | - Ähnlich wie Hennekam-Syndrom, aber mildere Ausprägung | Mechanosensitiver Ionenkanal, der über mechanische Reize eine Rolle in Lymphangiogenese spielt | ||
| EPHB4 | Lymphatisch-assoziierter fetaler Hydrops (LRFH) | 617300 | - Hydrops fetalis | Rolle im Remodeling der Lymphgefäße und der Klappenentwicklung | ||
| - Atriumseptumdefekt | ||||||
| - Varikosen | ||||||
| - Lymphödem der unteren Extremität | ||||||
| SOX18 | Hypotrichose-Lymphödem-Teleangiektasien-(renales) Syndrom | 607823 | - Lymphödem der unteren Extremität | Induziert Expression des lymphatischen Master-Transkriptionsfaktors PROX1 (induziert Entwicklung von LEZs aus venösen Endothelzellen) | ||
| - Hydrops fetalis | ||||||
| - Hydrozele | ||||||
| - Teleangiektasien | ||||||
| - Evtl. renale Beteiligung | ||||||
| PTPN14 | Choanalatresie-Lymphödem | 613611 | - Bilaterale Chonalatresie | Spielt eine Rolle in der Regulation der Lymphangiogenese und der Entwicklung der Choanen | ||
| - Lymphödem der unteren Extremität | ||||||
| DCHS1 | Van Maldergem Syndrom | 601390 | - Überlappende Klinik und Pathophysiologie mit dem Hennekam-Syndrom, jedoch ist ein Lymphödem beim Van Maldergem Syndrom selten | FAT4 und DCHS1 spielen gemeinsam eine Rolle bei der Entwicklung des Lymphgefäßsystems und in der Ausbildung von Lymphgefäß-Klappen | ||
| FLT4 | Milroy Erkrankung | 153100 | Kongenitales Lymphödem (Gruppe 2c) | - Lymphödem der unteren Extremität (im Besonderen Fußrücken und unterhalb des Knies) | FLT4 kodiert für den Vascular Endothelial Growth Factor Receptor 3 (VEGFR‑3), der auf LEzs exprimiert wird, an den VEGF‑C als wichtiger Mediator der Lymphangiogenese bindet [vgl. Schulte-Merker S, Sabine A, Petrova TV. Lymphatic vascular morphogenesis in development, physiology, and disease. Journal of Cell Biology, 2011; 193 (4): 607–618. doi: 10.1083/jcb.201012094. PMID: 21576390; PMCID: PMC3166860 und Kurek KC et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. American Journal of Human Genetics, 2012; 90 (6): 1108–1115] | |
| - Varikosen | ||||||
| VEGFC | Milroy-ähnliche Erkrankung | 615907 | - Positive Familienanamnese | |||
| FOXC2 | Lymphödem-Distichiasis-Syndrom | 153400 | Late-Onset Lymphödem (Gruppe 2d) | - Distichiasis | Transkriptionsfaktor, der eine Rolle in der Entwicklung der Klappen in Venen und Lymphgefäßen spielt | |
| - Lymphödem der unteren Extremität | ||||||
| - Varikosen | ||||||
| GJC2 | Vier-Extremitäten Lymphödem | 613480 | - Lymphödem der unteren Extremität, oberen Extremität und Genitalien | Connexin 47 spielt als Bestandteil von Gap-Junctions in Lymphgefäß-Klappen eine Rolle in der korrekten Ausbildung ebendieser | ||
- Entwicklung eines zum physiologischen Gangbild kongruenten polyzentrischen Knieorthesengelenks – Zwischenstand eines Forschungsprojektes — 10. April 2026
- Mikroprozessorgesteuerte stand- und schwungphasenkontrollierte Ganzbeinorthesen (SSCO): Entwicklung, Systeme und Versorgungsrealität in der orthopädietechnischen Praxis — 9. April 2026
- Gips oder Orthese bei distalen Radiusfrakturen? — 8. April 2026
- Schulte-Merker S, Sabine A, Petrova TV. Lymphatic vascular morphogenesis in development, physiology, and disease. Journal of Cell Biology, 2011; 193 (4): 607–618. doi: 10.1083/jcb.201012094. PMID: 21576390; PMCID: PMC3166860
- Brakenhielm E, Alitalo K. Cardiac lymphatics in health and disease. Nature Reviews Cardiology, 2019; 16 (1): 56–68. doi: 10.1038/s41569-018‑0087‑8. PMID: 30333526
- Potente M, Mäkinen T. Vascular heterogeneity and specialization in development and disease. Nature Reviews Molecular Cell Biology, 2017; 18 (8) :477–494. doi: 10.1038/nrm.2017.36. Epub 2017 May 24. PMID: 28537573
- Mortimer PS, Rockson SG. New developments in clinical aspects of lymphatic disease. Journal of Clinical Investigation, 2014; 124 (3): 915–921. doi: 10.1172/JCI71608. Epub 2014 Mar 3. PMID: 24590276; PMCID: PMC3938261
- Levick JR, Michel CC. Microvascular fluid exchange and the revised Starling principle. Cardiovascular Research, 2010 15; 87 (2): 198–210. doi: 10.1093/cvr/cvq062. Epub 2010 Mar 3. PMID: 20200043
- Rockson SG, Keeley V, Kilbreath S, Szuba A, Towers A. Cancer-associated secondary lymphoedema. Nature Reviews Disease Primers, 2019 28; 5 (1): 22. doi: 10.1038/s41572-019‑0072‑5. PMID: 30923312
- Connell FC, Gordon K, Brice G, Keeley V, Jeffery S, Mortimer PS, Mansour S, Ostergaard P. The classification and diagnostic algorithm for primary lymphatic dysplasia: an update from 2010 to include molecular findings. Clinical Genetics, 2013; 84 (4): 303–314. doi: 10.1111/cge.12173. Epub 2013 Jun 27. PMID: 23621851
- Mansour S, Martin-Almedina S, Ostergaard P. Genetic Disorders of the Lymphatic System. In: Pyeritz RE, Korf BR, Grody WW (Hrsg.): Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics. 7. Auflage. New York: Academic Press, 2020: 231–249
- Dempsey E, Homfray T, Simpson JM, Jeffery S, Mansour S, Ostergaard P: Fetal hydrops – a review and a clinical approach to identifying the cause. Expert Opinion on Orphan Drugs, 2020; 8 (2–3): 51–66
- Connell FC et al. The classification and diagnostic algorithm for primary lymphatic dysplasia: an update from 2010 to include molecular findings. Clinical Genetics, 2013; 84 (4): 303–314
- Gordon K et al. Update and audit of the St George’s classification algorithm of primary lymphatic anomalies: a clinical and molecular approach to diagnosis. Journal of Medical Genetics, 2020; 57 (10): 653–659
- Mansour S, Martin-Almedina S, Ostergaard P. Genetic Disorders of the Lymphatic System. In: Pyeritz RE, Korf BR, Grody WW (Hrsg.): Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics. 7. Auflage. New York: Academic Press, 2020: 231–249
- Dempsey E, Homfray T, Simpson JM, Jeffery S, Mansour S, Ostergaard P: Fetal hydrops – a review and a clinical approach to identifying the cause. Expert Opinion on Orphan Drugs, 2020; 8 (2–3): 51–66
- Gordon K et al. Update and audit of the St George’s classification algorithm of primary lymphatic anomalies: a clinical and molecular approach to diagnosis. Journal of Medical Genetics, 2020; 57 (10): 653–659
- Kemper C, Danyel M, Ott C‑E, Hägerling R. Genetik und Diagnostik des primären Lymphödems. Phlebologie, 2021; 50 (2): 105–114
- Ott C‑E, Danyel M, Kemper C, Hägerling R. Genetische Diagnostik des primären Lymphödems. Lymphologie in Forschung und Praxis, 2021; 25 (1) :14–20
- Mansour S, Martin-Almedina S, Ostergaard P. Genetic Disorders of the Lymphatic System. In: Pyeritz RE, Korf BR, Grody WW (Hrsg.): Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics. 7. Auflage. New York: Academic Press, 2020: 231–249
- Zschocke J. Pathomechanismen genetischer Krankheiten. Basiswissen Humangenetik. Berlin, Heidelberg: Springer, 2018: 67–95
- Kurek KC et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. American Journal of Human Genetics, 2012; 90 (6): 1108–1115
- Cohen MM, Jr. Proteus syndrome review: molecular, clinical, and pathologic features. Clinical Genetics, 2014; 85 (2): 111–119
- Rodriguez-Laguna L et al. CLAPO syndrome: identification of somatic activating PIK3CA mutations and delineation of the natural history and phenotype. Genetics in Medicine, 2018; 20 (8): 882–889
- Alitalo K. The lymphatic vasculature in disease. Nature Medicine, 2011; 17 (11): 1371–1380
- Connell FC, Gordon K, Brice G, Keeley V, Jeffery S, Mortimer PS, Mansour S, Ostergaard P. The classification and diagnostic algorithm for primary lymphatic dysplasia: an update from 2010 to include molecular findings. Clinical Genetics, 2013; 84 (4): 303–314. doi: 10.1111/cge.12173. Epub 2013 Jun 27. PMID: 23621851
- Morgan T. Turner syndrome: diagnosis and management. American Family Physician, 2007; 76 (3): 405–410
- Schaaf CP. Angeborene Fehlbildungssyndrome. Basiswissen Humangenetik. Berlin, Heidelberg: Springer, 2018: 241–262
- Mansour S, Martin-Almedina S, Ostergaard P. Genetic Disorders of the Lymphatic System. In: Pyeritz RE, Korf BR, Grody WW (Hrsg.): Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics. 7. Auflage. New York: Academic Press, 2020: 231–249
- Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. The Lancet, 2013; 381 (9863): 333–342
- Mansour S, Martin-Almedina S, Ostergaard P. Genetic Disorders of the Lymphatic System. In: Pyeritz RE, Korf BR, Grody WW (Hrsg.): Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics. 7. Auflage. New York: Academic Press, 2020: 231–249
- Paznekas WA et al. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Human Mutation, 2009; 30 (5): 724–733
- Brice G, Ostergaard P, Jeffery S, Gordon K, Mortimer P, Mansour S. A novel mutation in GJA1 causing oculodentodigital syndrome and primary lymphoedema in a three generation family. Clinical Genetics, 2013; 84 (4): 378–381
- Ostergaard P et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Natural Genetics, 2011; 43 (10): 929–931
- Mansour S, Martin-Almedina S, Ostergaard P. Genetic Disorders of the Lymphatic System. In: Pyeritz RE, Korf BR, Grody WW (Hrsg.): Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics. 7. Auflage. New York: Academic Press, 2020: 231–249
- Hägerling R et al. A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. The EMBO Journal, 2013; 32 (5): 629–644
- Alders M et al. Mutations in CCBE1 cause generalized lymph vessel dysplasia in humans. Natural Genetics, 2009; 41 (12): 1272–1274
- Fotiou E et al. Novel mutations in PIEZO1 cause an autosomal recessive generalized lymphatic dysplasia with non-immune hydrops fetalis. Nature Communications, 2015; 6: 8085
- Mansour S, Martin-Almedina S, Ostergaard P. Genetic Disorders of the Lymphatic System. In: Pyeritz RE, Korf BR, Grody WW (Hrsg.): Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics. 7. Auflage. New York: Academic Press, 2020: 231–249
- Martin-Almedina S, Mortimer PS, Ostergaard P. Development and physiological functions of the lymphatic system: insights from human genetic studies of primary lymphedema. Physiological Reviews, 2021; 101 (4): 1809–1871
- Martin-Almedina S, Mortimer PS, Ostergaard P. Development and physiological functions of the lymphatic system: insights from human genetic studies of primary lymphedema. Physiological Reviews, 2021; 101 (4): 1809–1871
- Gordon K et al. Mutation in vascular endothelial growth factor‑C, a ligand for vascular endothelial growth factor receptor‑3, is associated with autosomal dominant milroy-like primary lymphedema. Circulation Research, 2013; 112 (6): 956–960
- Mansour S, Martin-Almedina S, Ostergaard P. Genetic Disorders of the Lymphatic System. In: Pyeritz RE, Korf BR, Grody WW (Hrsg.): Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics. 7. Auflage. New York: Academic Press, 2020: 231–249
- Gordon K et al. Update and audit of the St George’s classification algorithm of primary lymphatic anomalies: a clinical and molecular approach to diagnosis. Journal of Medical Genetics, 2020; 57 (10): 653–659
- Mansour S, Brice GW, Jeffery S, Mortimer P. Lymphedema-Distichiasis Syndrome. In: Adam MP et al. (Hrsg.). GeneReviews(®). Seattle: University of Washington, 2005 (updated 2019)
- Gordon K et al. Update and audit of the St George’s classification algorithm of primary lymphatic anomalies: a clinical and molecular approach to diagnosis. Journal of Medical Genetics, 2020; 57 (10): 653–659
- Mansour S, Martin-Almedina S, Ostergaard P. Genetic Disorders of the Lymphatic System. In: Pyeritz RE, Korf BR, Grody WW (Hrsg.): Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics. 7. Auflage. New York: Academic Press, 2020: 231–249
- Ferrell RE et al. GJC2 missense mutations cause human lymphedema. American Journal of Human Genetics, 2010; 86 (6): 943–948
- Alpaslan M et al. Ureteropelvic junction obstruction with primary lymphoedema associated with CELSR1 variants. Journal of Medical Genetics, 2023; jmg-2023–109171
- Hägerling R et al. VIPAR, a quantitative approach to 3D histopathology applied to lymphatic malformations. JCI Insight, 2017; 2 (16): e93424
- Lampejo AO, Ghavimi SAA, Hägerling R, Agarwal S, Murfee WL. Lymphatic/blood vessel plasticity: motivation for a future research area based on present and past observations. American Journal of Physiology-Heart and Circulatory Physiology, 2023; 324 (1): H109–H121
